医薬品情報管理学[4]
医薬品情報21
古泉秀夫
| 情報源としてのインタビューフォーム |
1]Interview Formの概要
Interview Form(以下「IF」と略す)は、当初、病院において医薬品情報を担当する薬剤師が、製薬企業の医薬品情報担当者(以下「MR」と略す)と面談し、情報を蒐集する際、聞き取り調査をするための項目チェックを目的として作成されたものである。しかし、実際には、項目が細分化するにつれて、MRの対応が大変だということで、製薬企業がそれぞれ印刷して配布するようになったという経緯がある。
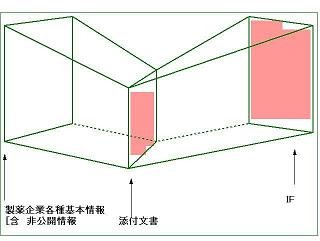
図7.添付文書とIFの相関図
添付文書は、薬事法の規定があり、膨大な情報を圧縮するという作業が必要になるが、IFには紙数の制約はなく、また、記載する内容についても、出典が明示される限り、何等制約は設けられていない。つまりIFは、添付文書に収載できない各種情報を詳細に収載できるということで、添付文書に収載された情報の投影図であるとすることが出きる。
2]IFの問題点
本来であれば、添付文書とIFを蒐集することによって、その医薬品の基本的情報の80%程度が蒐集可能であるという状況が必要であるが、実際には添付文書の焼き直し程度のIFを発行している製薬企業もあり、何の目的で発行したのか意味不明な内容のものもある。更に記載されている項目別に詳細に検討すると、書き込み不足と思われる部分が多い。製薬企業もIFに対する認識を変え、添付文書では出来ない情報を提供する媒体として明確に位置付けるべきである。IFの情報内容が充実すれば、基本的な情報について、一々製薬企業に問い合わせる必要が無くなり、製薬企業側にとっても、大きな利益となるはずである。
| 項目 | 添付文書 | IF |
| 記載内容の法的規制 | 有 | 無 [ただし、出典明記] |
| 記載内容の量的規制 | 有 | 無 |
| 情報の優位性 | 第1位 | 補助的 |
| 情報の詳細性 | 限界 | 無限界 |
| 変更情報の確保 | 即応性 | 非即応性 |
| 収載項目 | 法規制 | 日病薬IF委員会設定 |
| 情報の範囲 | 限界 | 無限界 |
| 情報の深度 | 限界 | 無限界(文献的裏付) |
| 改訂の即応性 | 速 | 遅 |
なお、添付文書とIFの記載項目を比較検討すると、表4.の通りである。
I.概要に関する項目
| 記載項目 | IF | 添付文書 |
| 1. 開発の経緯 | ○ | 改訂新文書(削除) |
| 2.製剤の特徴及び有用性、類似薬との比較 | ○ | ? |
| 3.主な外国での発売状況 | ○ | ? |
II.名称に関する項目
| 記載項目 | IF | 添付文書 |
| 4.商品名 | ○ | ○ |
| 5.一般名 | ○ | ○ |
| 6.構造式又は示性式 | ○ | ○ |
III.原薬の性状に関する項目
| 記載項目 | IF | 添付文書 |
| 7.原薬の規制区分 | ○ | ○ |
| 8.起 源 | ○ | ? |
| 9.物理化学的性状 | ○ | ○ |
| 10.原薬の安定性 | ○ | △ |
| 11.原薬の確認試験法 | ○ | ? |
| 12.原薬の純度試験法 (定量法) | ○ | ? |
| 13.構造上関連ある化合物又は化合物群 | ○ | ? |
IV.製剤に関する項目
| 記載項目 | IF | 添付文書 |
| 14.剤 形 | ○ | ○ |
| 15.製剤上の特徴 | ○ | △ |
| 16.製剤の組成 | ○ | ○ |
| 17.製剤の安定性 | ○ | △ |
| 18.他剤との配合変化 | ○ | △ |
| 19.混入する可能性のある夾雑物 | ○ | ? |
| 20.製剤中の原薬確認試験 | ○ | ? |
| 21.製剤中の原薬定量法 | ○ | ? |
| 22.容器の材質 | ○ | ? |
| 23.その他 | ○ | ? |
V.治療に関する項目
| 記載項目 | IF | 添付文書 |
| 24.効能・効果 | ○ | ○ |
| 25.用法・用量 | ○ | ○ |
| 26.臨床適用 | ○ | ○ |
| 27.その他の薬理作用 | ○ | ○ |
| 28.診断的特徴 | ○ | ○ |
VI.使用上の注意に関する項目
| 記載項目 | IF | 添付文書 |
| 29."警告"とその理由 | ○ | ○ |
| 30."一般的注意"とその理由及び処置方法 | ○ | ○ |
| 31."禁忌"とその理由 | ○ | ○ |
| 32."慎重投与"とその理由 | ○ | ○ |
| 33.副作用 | ○ | ○ |
| 34.薬物アレルギーに対する注意・試験法 | ○ | ? |
| 35.高齢者への使用に関する注意 | ○ | ○ |
| 36.妊婦等への使用に関する注意 | ○ | ○ |
| 37.授乳婦への使用に関する注意 | ○ | ○ |
| 38.未熟児、新生児、乳児、幼児、小児への使用に関する注意 | ○ | ○ |
| 39.相互作用 | ○ | ○ |
| 40.臨床検査値への影響 | ○ | ○ |
| 41.適用上の注意 | ○ | ○ |
| 42.薬剤交付時の注意事項 | ○ | ○ |
| 43.過量投与時の処置 | ○ | ○ |
| 44.腎障害時の投与 | ○ | ○ |
| 45.肝障害時の投与 | ○ | ○ |
| 46.その他 | ○ | ? |
VII.薬効薬理に関する項目
| 記載項目 | IF | 添付文書 |
| 47.薬理学的に関連ある化合物・化合物群 | ○ | ? |
| 48.薬理作用 | ○ | ○ |
| 49.薬理学的特徴 | ○ | ○ |
VIII. 体内薬物動態に関する項目
| 記載項目 | IF | 添付文書 |
| 50.血中濃度の推移・測定法 | ○ | △ |
| 51.薬物速度論パラメータ | ○ | ○ |
| 52.作用発現時間 | ○ | △ |
| 53.作用持続時間 | ○ | △ |
| 54.吸収 | ○ | ○ |
| 55.分布 | ○ | ○ |
| 56.代謝 | ○ | ○ |
| 57.排泄 | ○ | ○ |
| 58.透析等による除去率 | ○ | △ |
IX.非臨床試験に関する項目
| 記載項目 | IF | 添付文書 |
| 59.一般薬理 | ○ | ○ |
| 60.毒性 | ○ | ? |
| 61.動物での体内動態 | ○ | ○ |
X. 取扱い上の注意、包装、承認等に関する項目
| 記載項目 | IF | 添付文書 |
| 62.有効期限又は使用期限 | ○ | ○ |
| 63.貯法・保存条件 | ○ | ○ |
| 64.薬剤取扱い上の注意点 | ○ | ○ |
| 65.包装・単位 | ○ | ○ |
| 66. 同一成分、同効薬 | ○ | ? |
| 67.製造・輸入承認年月日、承認番号 | ○ | ○ |
| 68.薬価基準収載年月日 | ○ | ○ |
| 69. 再審査期間の年数 | ○ | ○ |
| 70.長期投与の可否 | ○ | ○ |
| 71.厚生省薬価基準収載医薬品コード | ○ | ? |
| 72.国際誕生日 | ○ | ○ |
XI.文献
| 記載項目 | IF | 添付文書 |
| 73.引用文献 | ○ | ○ |
| 74.その他の参考文献 | ○ | ? |
| 75.文献請求先 | ○ | ○ |
| 76.問い合わせ先 | ○ | ○ |
| 77. 会社名及び住所 | ○ | ○ |
表4.の各項目は、IF上に設定されている項目である。添付文書欄の○印は、添付文書上に記載されている項目であり、△印は添付文書上に記載されているが、情報源としては、不十分な内容と判断されるものである。空欄は当然のこととして添付文書上には記載されていない情報であることを示す。
3]新たに追加が求められる情報
上記以外にIF上に更に追加が求められる情報として、次のものが上げられる。
- 血管外漏出時の処置:添付文書上に点滴静注時に血管外に漏出した場合、組織障害が起こるの記載がされているが、具体的な処置方法の記載がされていないため、より具体的な処置方法の記載が求められる。
- 定期的な検査の具体的間隔:重篤な副作用防止のために、『定期的な検査』の必要性が添付文書上に記載されているが、定期的の具体的な頻度の記載がされておらず、投与初期にはどの程度の間隔で実施するのか、長期投与の際の検査間隔等の具体的記載がされていない。患者の安全性確保のために必要とされる重要な情報であり、より具体的な記載が必要である。
- 手術前後の投与上の注意:添付文書の一部には記載されているが、技術的に進歩している現在の手術でも、同様な注意が必要なのかどうか。また必要とすれば、より具体的な処置方法の記載が求められる。
- 他臓器障害時の投与上の注意:腎障害時の投与については、具体的な記載が見られるが、肝障害等の多臓器障害時の投与方法についてより具体的な情報。
- 妊婦への投与:表記が曖昧であり、FDA評価等の具体例を参照して、実務に役立つ評価法を明示すべきである。
- 授乳婦への投与:添付文書上の記載は、具体性のない安易な記載がされており、具体的に臨床現場で役立つ情報を記載
等が求められる。
更に製薬企業は、社内資料として、未公開を前提条件とする資料を持参することがあるが、公表できない情報は情報ではない。最低限IFに収載可能な情報として精度の高い情報にまで高めるべきであり、臨床現場が必要とするより実務的な情報作成の基本姿勢を示すべきである。
- 古泉秀夫:医薬品情報管理学[3];THPA,45(1):13-21(1996)